
Amyotrofisk lateral sklerose (ALS)
Amyotrofisk lateral sklerose – ALS er en progressiv sygdom, der hører til i gruppen af neurodegenerative sygdomme, der også betegnes motorneuron sygdomme (MND), og som ud over klassisk ALS inkluderer progresiv bulbær parese (bulbær ALS), progressiv muskelatrofi (PMA) og ALS/FTD
Primær lateral sklerose (PLS) betegnes også som en motorneuron sygdom, men er ikke en ALS-undertype.
ALS angriber både 1. og 2. neuron
ALS angriber de neuroner (nerveceller) i hjerne og rygmarv, som styrer den tværstribede muskulatur.
Man inddeler de motoriske neuroner i 1.neuron (øvre), og 2. neuron (nedre).
Første neuron betegner de motoriske neuroner, der ligger i motorisk kortex (hjernebarken), hvor aksonet (nervetråden) løber ned til et nyt motorisk neuron (2. neuron) beliggende enten i medulla oblongata (forlængede marv) eller i medulla spinalis (rygmarven) forreste del, som kaldes forhornet (2.neuronet i forhornet benævnes også forhornscelle).
Andet neuron sender et langt akson ud til skeletmuskulatur i hele kroppen, herunder også respirationsmusklerne (vejrtrækningsmuskler).
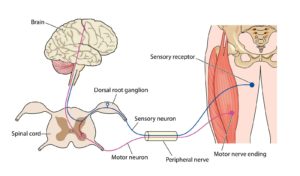
Ved klassisk ALS og bulbær ALS er både 1. og 2. motorneuron påvirket. Ved PMA er 2. motorneuron påvirket. Ved ALS/FTD er nervecellerne i frontal- og/eller temporallapperne påvirket udover påvirkning af én eller begge motoriske neuroner.
-
Forekomst
Der er i Danmark cirka 400 personer med ALS, og der diagnosticeres ca. 150 nye tilfælde om året.
ALS er en sygdom, der hyppigst rammer i aldersgruppen over 50 år, men den forekommer ned til 20-årsalderen og også op i 90’erne. Lidt flere mænd end kvinder får ALS.
Årsag
Ved ALS betragtes de underliggende sygdomsmekanismer som værende multifaktorelle, hvor både genetik og udefrakommende påvirkninger spiller ind.
De fleste tilfælde forekommer sporadisk (tilfældigt), men hos en undergruppe af patienterne forekommer sygdommen familiært. I dag kendes gener, som er associeret med både familiær ALS (FALS) og sporadisk ALS (SALS).
Aktuelt har man kendskab til mere end 25 gener, som mistænkes for at spille en rolle ift. udvikling af ALS, og antallet vokser vedvarende. Der forskes intensivt i de allerede fundne genmutationers betydning samt i at identificere andre potentielle kausale mutationer. De to hyppigst forekommende mutationer er nogle af første, der blev opdaget.
Det anslås, at ca. 10 % af ALS-tilfælde er familiært betingede. Den enkelte specifikke genetiske mutation kan i samme familie forårsage meget forskellige kliniske fænotyper. Der kan således ses et bredt spektrum af diagnoser (ALS, frontotemporal demens og Parkinson sygdom) med umiddelbart samme genetiske fejlkode.
De to hyppigste gener involveret i ALS:
- C9orf72 er det gen, hvori der hyppigst findes mutationer i både FALS og SALS. Arvegangen er autosomal dominant. Mutationen findes hos ca. 40 % med FALS og hos ca. 10 % med SALS. Hvis fænotypen i familien er ALS hos nogle, FTD hos andre eller en fænotype med blandet ALS og FTD påvises mutation hos helt op mod 80 %. Fænotypen kan også være med parkinsonisme. Penetransen (antal af personer med mutationen, der får sygdom) vurderes at være høj, men ikke 100 %.
- I SOD1-genet er der beskrevet mange forskellige sygdomsfremkaldende mutationer. Arvegangen er hyppigst autosomal dominant, men en hyppig missense-mutation i den skandinaviske befolkning, p.Asp90Ala, medfører en autosomal recessiv arvelig form, hvor progressionshastigheden er langsommere. SOD1-mutationer identificeres hos 15-20 % med FALS, og hos 2-7 % med SALS. Fænotypen ses sjældnere med kognitiv påvirkning. Penetransen vurderes at være høj, men ikke 100 %.
Reference: Neurologisk national behandlingsvejledning – ALS og genetik
-
ALS-diagnosen kan i starten være meget vanskelig at stille. Det skyldes, at der ikke findes et enkelt sygdomstegn eller en enkelt prøve/test (fx blodprøve), der giver en entydig diagnosticering af sygdommen. Derfor har nogle personer med ALS været igennem et langt udredningsforløb, inden de får stillet diagnosen.
ALS-diagnosen stilles, ved at der kan påvise både 1. og 2.motorneurons symptomer i mindst 1 region af kroppen eller 2. motorneurons symptomer i mindst 2 regioner af kroppen. For ALS gælder, at symptomer ikke kan forklares ved anden sygdom. Samtidig må der ikke være påvirkning af følesansen eller andre sanser. Der skal være progredierende/fremadskridende motoriske udfald dokumenteret ved klinisk undersøgelse eller anamnese forudgået af normal motorisk funktion.
ALS er en klinisk diagnose med forværring over tid. For at udelukke andre sygdomme i nervesystemet, kan der foretages en række undersøgelser, som kan bekræfte/understøtte ALS-diagnosen eller afkræfte denne.
- Blodprøver: foretages rutinemæssigt
- Genetisk undersøgelse: kan være relevant, hvis der har været flere tilfælde i familien.
- Rygmarvsvæskeundersøgelse: kan udelukke andre årsager og være med til at påvise 1. neurons påvirkning.
- MR- eller CT-scanning af hjerne og rygmarv: Kan udelukke andre diagnoser, hvor tryk på nervebaner kan forekomme.
- Neurofysiologisk undersøgelse i form af EMG (elektromyografi), ENG (elektroneurografi), MEP (motor evoked potential) og SSEP (Somato-Sensorisk-Evokerede-Potentialer). Ved EMG måles den elektriske aktivitet i musklerne, der ved ALS viser et bestemt mønster med denervering og evt. Ved ENG måles nerveledningshastigheder i de motoriske nerver (2.neuron), og ved MEP måles, hvordan 1. neuron fungerer. Ved SSEP er formålet at undersøge de sensoriske nervebaner. Den neurofysiologiske undersøgelse kan være med til at understøtte den objektive neurologiske undersøgelse.
-
Symptomer ved sygdomsdebut
- 2/3 af MND-patienter debuterer med ekstremitetssymptomer
- 1/3 debuterer med bulbære symptomer (tale-, tygge og synkebesvær)
- Få procent debuterer med frontotemporal demens (FTD)
- Sjældent, men forekommende, ses debut med vejrtrækningsbesvær
Symptomer, der opstår i forløbet
Sygdommen kan således debutere med forskellige symptomer, men som sygdommen udvikler sig, vil de fleste opleve en stor del af nedenstående symptomer:
- Asymmetriske pareser (muskelslaphed eller lammelser)
- fascikulationer (sitren i musklerne)
- tab af muskelkraft, muskelatrofi (nedsat muskelfylde)
- hyperrefleksi/arefleksi (påvirkede reflekser)
- dysfagi (synkebesvær)
- dysartri (talepåvirkning)
- hos op mod 50 % ses kognitiv dysfunktion (påvirkning af tankeprocesser som fx refleksion og overblik) og personlighedsforandring. Hos op til 10 % udvikles frontotemporal demens (FTD) Læs mere nedenfor
I ca. en tredjedel af tilfældene debuterer ALS med såkaldte bulbære symptomer, dvs. symptomer, der knytter sig til muskulaturen i ansigtet, tungen og svælget. Symptomerne i det bulbære område starter ofte med lette taleforstyrrelser (fx hæshed, eller lav stemme), der ofte i begyndelsen kun bemærkes af personen selv eller dennes nærmeste, især ved træthed.
Der kan ses dysfagi. Munden, svælget og kindernes muskler påvirkes, hvilket medfører nedsat evne til at tygge maden med tendens til fejlsynkning. Øget spytflåd er desuden en hyppig følge af den bulbære påvirkning.
Uprovokerede muskelsmerter er et hyppigt symptom. Smerterne kan være et tidligt symptom, men de kan også støde til senere i forløbet. De kan skyldes påvirkning af smertereceptorer i musklerne, men i andre tilfælde kan man ikke forklare smerterne. Efterhånden som muskelkraften påvirkes, kan der også ses ledsmerter på baggrund af overbelastning og kontrakturer.
Referencer:
Neurologisk national behandlingsvejledning – diagnostik og klassifikation af motor neuron sygdomme
-
Sygdommen udvikler sig meget forskelligt fra person til person, men altid gradvist. Symptomerne er jævnt progredierende både i sværhedsgrad og antallet af involverede områder. Efterhånden som sygdommen udvikler sig, vil også respirationsmusklerne påvirkes.
Ved de bulbære symptomer kan der hos nogle ses tab af emotionel kontrol. Dette kan føre til umotiveret og ukontrolleret gråd og/eller latter, hvilket kan opleves som socialt hæmmende og pinagtig både for personen selv, og for familie og øvrige omgivelser. Denne tilstand kan ofte behandles effektivt med en lille dosis antidepressiv medicin.
-
Undersøgelser viser, at op mod 50 % af dem, der diagnosticeres med ALS har, eller vil udvikle, ændret adfærd og personlighed. Hos ca. 10 % ses der udvikling af frontotemporal demens (FTD). Studier tyder på, at op mod 80 % med ALS i slutningen af sygdomsforløbet vil vise tegn på frontotemporal dysfunktion.
Hvordan viser de kognitive forandringer sig?
Den ændrede adfærd kan være vanskelig at bemærke for neurologen, da de kun har mulighed for kortvarig observation. Det er derfor ofte de pårørende, der i de hjemlige omgivelser bemærker, at personen med ALS har ændret adfærd.
Ved en neuropsykologisk undersøgelse kan man få et indtryk af, om der er kognitive forandringer. Det er forskelligt, hvornår i forløbet de neurologiske afdelinger henviser til en sådan undersøgelse. Nogle afdelinger har denne undersøgelse som en del af det basale udredningsprogram, og andre henviser først senere i forløbet.
Ved frontotemporal dysfunktion ses ofte
- en ændret adfærd i form af en øget impulsiv og uovervejet adfærd
- en ligegyldighed overfor sociale normer
- en påvirket/manglende empati overfor andre mennesker.
Ofte nævnte eksempler på en sådan adfærd kunne være; upassende bemærkninger, eller fx tendens til at ”proppe” maden i munden (også på trods af evt. spise- synkeproblemer). Kognitive forandringer kan vise sig ved vanskeligheder med at bevare overblik og træffe rationelle beslutninger.
Hos nogle ses, at præmorbide personlighedstræk forstærkes som f.eks. stædighed, opfarenhed og rastløshed. Der ses ofte også påvirkning af selvindsigten, manglende sygdomserkendelse, samt manglende hæmning og situationsfornemmelse.
Symptomerne kan være vanskelige at skelne fra en forventelig væremåde grundet fx nedsat funktionsniveau og kommunikationsevne (ved påvirket taleevne), eller evt. en depressionstilstand.
Adfærdsændringer og de kognitive ændringer forværres over tid, ligesom de fysiske symptomer ved ALS.
Frontotemporal demens (FTD)
Hvis personen med ALS har udtalte symptomer af ovenstående type, dvs. både ændret adfærd, kognition samt sprogforstyrrelser, kaldes tilstanden for frontotemporal demens (FTD). I modsætning til andre former for demens er hukommelsen ikke nødvendigvis påvirket, men der kan ses sproglige forstyrrelser.
FTD kan optræde som debutsymptom ved ALS, og de pårørende kan således have oplevet adfærdsændringer op til flere år før de motoriske symptomer optræder og diagnosen stilles. FTD kan dog også udvikle sig senere i ALS-sygdomsforløbet efterfølgende de motoriske symptomer. FTD forværres igennem sygdomsforløbet.
Når ALS-sygdommen kompliceres af FTD, er det erfaringen, at sygdomsforløbet ofte er hurtigere forløbende.
Der findes ingen medicinsk behandling, der kan bedre symptomer eller stoppe udviklingen af FTD, men information til de pårørende og hjælperne er vigtig.
Referencer:
ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS
Forventet levetid og den sidste tid
ADVARSEL! Er du eller din nærtstående ramt af ALS, gør vi opmærksom på, at nedenstående afsnit indeholder information om fremtidsudsigter og forventet levetid. Vi anbefaler, at du først læser nedenstående, når du føler dig klar til det.
Forventet levetid kan variere meget afhængigt af typen af ALS, og i hvilken grad du er påvirket af sygdommen.
-
Den gennemsnitlige levetid uden respirationshjælpemidler er 2-3 år fra diagnosetidspunktet, men variationen er stor.
Den seneste opgørelse i RCFM fra 2019 viste, at 15 % af personer med ALS er i live 3 år efter diagnosetidspunktet. Den bulbære debut form har oftest et hurtigere forløb end den spinale form.
-
Den syge bliver over tid sengeliggende og afhængig af hjælp til alle funktioner. Respirationen vil blive tiltagende svækket, og der vil være stigende risiko for lungeinfektioner. I takt med at respirationsmusklerne svækkes, vil der ske en gradvis CO2-ophobning, der i sidste ende medfører, at patienten bliver tiltagende komatøs og til sidst sover ind.
Det anbefales at inddrage de palliative specialister, når sygdommen er fremskreden, da de kan bidrage til den nødvendige lindrende behandling. Ofte vil det dog være en fordel at have stiftet bekendtskab med de palliative teams tidligere i forløbet, hvor der måske er mere ro til at lære familien at kende og dermed skabe gode forudsætninger for et godt forløb senere hen.
Hvis man har fået tilbudt og takket ja til respiratorbehandling (både invasiv og non-invasiv (NIV)), vil der ved behandlingens begyndelse være truffet en aftale om, hvornår behandlingen skal afsluttes, vel vidende, at ALS sygdommen i øvrigt vil progrediere uafhængigt af respirationshjælpemidlerne.
I alle tilfælde er det en lægelig beslutning, der træffes senest, når patienten ikke længere på nogen måde er i stand til at kommunikere, dvs. når der ikke er mere bevægelse i kroppen (det vil typisk være evnen til bevæge øjnene, der som det sidste ophører). Tilstanden kaldes locked-in.
-
Medicinsk behandling
Der findes i øjeblikket ingen behandling, hverken medicinsk eller kirurgisk, der kan stoppe eller helbrede ALS.
Riluzol er den første medicin, som har vist sig at have en positiv effekt på overlevelsestiden hos personer med ALS. Undersøgelser har vist, at den gennemsnitlige overlevelsestid for de patienter, der fik Riluzol, var signifikant længere end for dem, der fik placebo. Ved sammenlignelige undersøgelser over 18 måneder finder man, at patienter, der fik 100 mg Riluzol dagligt, i gennemsnit overlevede ca. 2 måneder længere end dem, der fik placebo.
50 mg Riluzol dagligt var ikke mere effektivt end placebo, og 200 mg dagligt var ikke mere effektivt end 100 mg dagligt. I de sene stadier af ALS virkede lægemidlet ikke bedre end placebo.
Der kan forekomme bivirkninger i form af kvalme, diarré og øget træthed.
Referencer:
Anden behandling
En del af symptomerne, der ledsager sygdommen, kan afhjælpes eller lindres, hvis man får den rigtige behandling eller de rigtige hjælpemidler.
Medicinsk behandling af spasticiteten kan være nødvendigt. Der kan anvendes spasmolytika (anti-spastika) som f.eks Baclofen eller neuromuskulært blokerende midler som Botolinumtoksin injektioner. Behandling af spasticitet kan dog være et tveægget sværd i de tilfælde, hvor der er udtalt kraftnedsættelse, og hvor det er spasticiteten, der bevarer gangfunktionen, idet muskelspændingen dermed nedsættes medførerende nedsat balance og øget faldrisiko.
Spyt og sekretproblemer kan behandles med antikolinergika som Scopoderm® og atropin, disse nedsætter spytsekretionen, men kan også bevirke, at slimet bliver mere sejt. Tricycliske antidepressiva, som amitriptylin, kan også anvendes eller injektioner med botulinumtoksin.
Respirationsproblemer kan lindres og til en vis grad afhjælpes med forskellige behandlinger og tiltag.
Ved udtalte dysfagisymptomer og vægttab kan den behandlende neurolog henvise til anlæggelse af en PEG-sonde.
Fysioterapeutisk behandling:
Spasticitet, som oftest forværres ved kulde, kan være meget generende og hæmme bevægelserne. Det er derfor vigtigt at foretage udspænding for at bevare mobiliteten samt undgå fejlstilling over de enkelte led. Fysisk aktivitet nedsætter desuden stivheden og kan dæmpe eventuelle muskelkramper.
Træning af de ikke-afficerede muskler er ligeledes vigtigt, for at undgå overbelastningsskader samt med henblik på at bevare et så højt funktionsniveau som muligt.
Se de kliniske retningslinjer for fysioterapi til patienter med ALS
Ergoterapeutisk behandling: til hjælp ift. dysfagi og i forhold til hjælpemidler.
Logopædisk behandling: ved eventuelle dysartri-gener.
Palliative teams: Når sygdommen er fremskreden, og der kommer et tiltagende behov for lindrende behandling, anbefales det at inddrage de palliative specialister.
Referencer:
Neurologisk national behandlingsvejledning for motorneuron sygdomme
Rettidig rehabilieringsindsats
Socialstyrelsen har i samarbejde med bl.a. RCFM udarbejdet en pjece, der beskriver vigtigheden af en rettidig indsats i forløb med hurtig progressions.
Det gode sagsforløb for voksne med hurtigt fremadskridende sygdomme
Relevante samarbejdspartnere
- ALS-teams på neurologiske afdelinger
- Respirationscentrene på AUH, OUH og RH
- Palliative specialister
- Kommunale støtteforanstaltninger
- Kommunikationscentre
Beskrivelse af motorneuronsygdom (MND)
ALS (Amyotrofisk lateral sklerose) er en sygdom, der er lokaliseret i de motoriske nerveceller, og de benævnes derfor overordnet motorisk nervesygdomme.
Den engelske betegnelse MND (Motor Neuron Disease) benyttes mere og mere, også i Danmark og af de danske neurologer. Betegnelsen MND dækker over en række karakteristiske, progredierende symptomer og kan inddeles i 4 hovedtyper. Sammensætningen af symptomer samt rækkefølgen de opstår i, kan være afgørende for, hvilken type MND der diagnosticeres.
De 4 hovedtyper er:
- Klassisk ALS
- Progressiv bulbær parese (Bulbær ALS)
- Progressiv muskelatrofi (PMA)
- ALS med Fronto Temporal Demens eller frontotemporal dysfunction (ALS/FTD eller ALS/FTDdys)
Fælles for de 4 typer er påvirkningen af de motoriske neuroner (nerveceller), der ved viljens hjælp styrer musklernes bevægelse, og som i takt med sygdommens progression gradvist går til grunde.
Hvilken af de 4 typer, der er tale om, afhænger af, hvilke motoriske neuroner der er påvirket. Man inddeler de motoriske neuroner i 1.neuron (øvre), og 2. neuron (nedre).
Første neuron betegner de motoriske neuroner, der ligger i den motoriske cortex (hjernebarken), hvor axonet (nervetråden) løber ned til et nyt motorisk neuron (2. neuron) beliggende enten i medulla oblongata (forlængede marv), eller i medulla spinalis (rygmarvens) forreste del, kaldet forhornet (2.neuronet i forhornet benævnes også forhornscelle).
Andet neuron sender et langt axon ud til skeletmuskulaturen i hele kroppen, herunder også respirationsmuskler (vejrtrækningsmuskler).
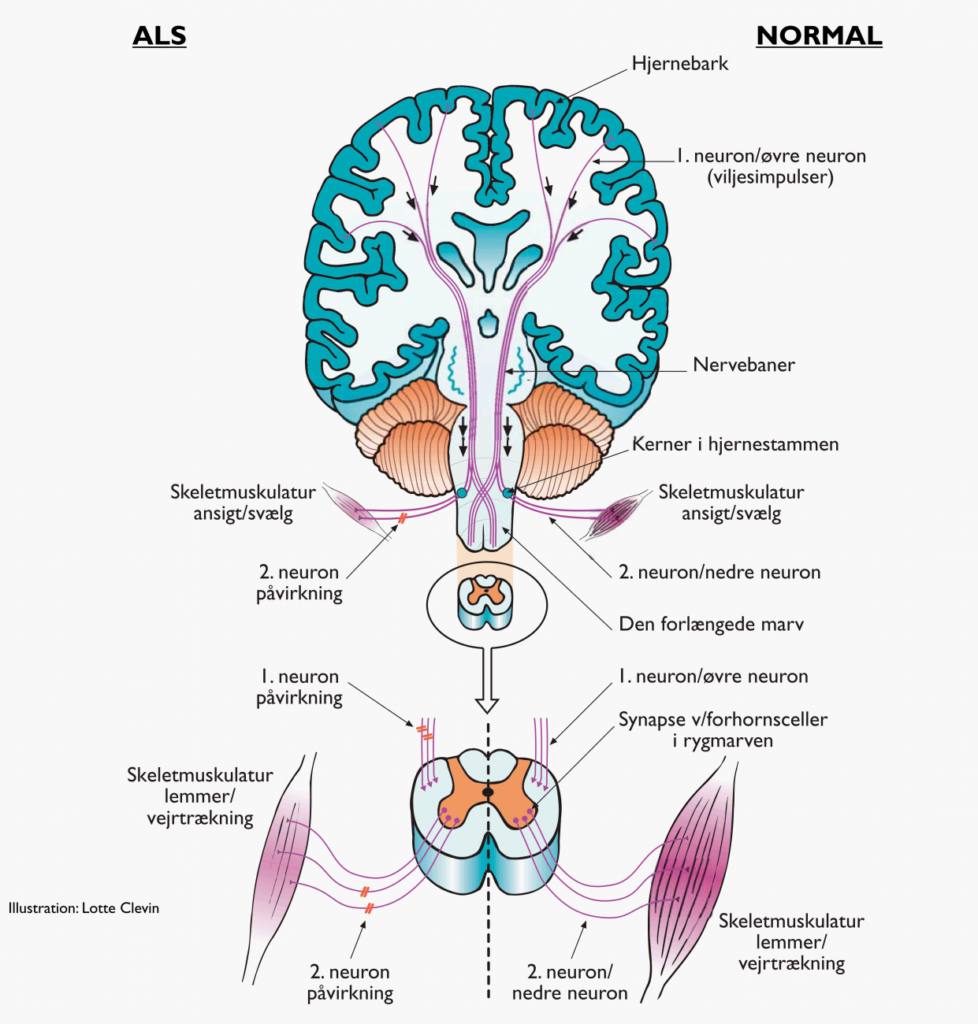
Ved klassisk ALS, Bulbær ALS og ALS/FTD er både 1. og 2. motorneuron påvirket i mindst en region af kroppen. Ved PMA er der alene påvirkning af 2. motoriske neuron i mindst to regioner af kroppen. For nærmere beskrivelse af symptomer ved de enkelte hovedtyper, se venligst sygdomsbeskrivelse for disse
Ødelæggelsen af neuronerne og axonerne kan således opstå og udvikle sig i hjernen og/eller i rygmarven. Afhængig af hvor påvirkningen starter, kan symptomerne vise sig i fx en arm eller svælget, eller mere udbredt i fx begge arme, eller arme og ben. Efterfølgende kan symptomerne sprede sig til resten af kroppens skeletmuskulatur. Sygdomsforløbet for den enkelte kan derfor være meget forskelligt, afhængigt af hvordan og hvor neurodegenerationen (nerveødelæggelse) og dermed symptomerne starter.
Tidligt i sygdomsforløbet kan det være vanskeligt at skelne mellem de forskellige typer, da der er overlappende symptomer.
Inddelingen af ALS-sygdommen i de 4 undertyper er et forsøg på at beskrive sygdommens forventede udvikling og prognose på diagnosetidspunktet. Det er vigtigt at bemærke, at nogle som først diagnosticeres som PMA senere i forløbet kan få ændret diagnosen til ALS i takt med at deres symptomer udvikler sig.


