
Charcot-Marie-Tooth sygdom (CMT)
CMT er en fællesbetegnelse for en gruppe af de arvelige motorsensoriske neuropatier, der også kaldes HMSN. Charcot, Marie og Tooth er navnene på de tre læger, der samtidigt, men uafhængigt af hinanden, først beskrev sygdommen i 1886. Sygdommen omtales oftest som CMT.
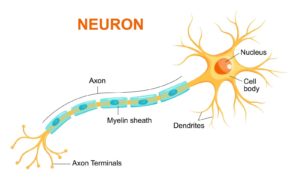
Sygdommen er progredierende og påvirker både de motoriske og sensoriske perifere nerver. Der kan være tale om påvirkning af enten selve aksonet eller nervernes myelinskeder.
Hvis sygdommen påvirker myelinskeden, reduceres ledningshastigheden, hvorved det tager længere tid for nervens signal at nå frem. Hvis sygdommen sidder i aksonerne, er ledingshastigheden normal, men signalets styrke er svagere. Da nervernes signaler til musklerne er påvirkede, vil der over tid udvikles muskelatrofi og kraftnedsættelse. Karakteristisk for sygdommen er, at det særligt er musklerne i fødder og hænder, der påvirkes.
Der findes forskellige typer og subtyper af CMT. Typen bestemmes typisk ved det kliniske billede, arvegangen, og hvilken del af nerven, der er afficeret (akson eller myelinskede). Subtype gives, hvis man kender den præcise genetiske årsag. Man taler om den demyeliniserende type, CMT1, den aksonale type, CMT2, en intermediær type, CMT4 og X-bundet CMT. Hver subtype har flere undertyper.
Sygdommen hører under gruppen af hereditære motorsensoriske neuropatier.
Hereditær motorsensorisk neuropati (HMSN)
Er en samlende betegnelse for de arvelige sygdomme hvor der sker påvirkning af både de motoriske og sensoriske perifere nerver. HMSN hører overordnet til gruppen af de arvelige neuropatier.
Man kan betragte gruppen af neuropatier som et spektrum, hvor der i den ene side findes de rent sensoriske neuropatier og i den anden ende de rent motoriske neuropatier. De arvelige neuropatier, hvor der er påvirkning af både motoriske og sensoriske nerver, kaldes samlet HMSN.
Der kan være tale om påvirkning af enten selv axonet eller nervernes myelinskeder. Hvis sygdommen påvirket myelinskeden, reduceres ledningshastigheden, hvorved at det tager længere tid for nervens signal at nå frem. Hvis sygdommen sidder i axonerne er ledingshastigheden normal, men signalets styrke er svagere.
I RCFM har vi tilknyttet brugere med CMT og HNPP, der begge er arvelige og progredierende lidelser. Genetisk minder sygdommene om hinanden, men det kliniske billede er ret forskelligt.
-
Forekomst
CMT er den hyppigst forekomne arvelige neuropati og forekommer hos 10-20 ud af 100.000
Den kan ramme både børn og voksne uafhængigt af køn, men der er en overhyppighed af mænd/drenge.
Den hyppigste form i Danmark er CMT type 1a.
Årsag til sygdommen
Sygdommen skyldes mutationer i de gener, der er ansvarlige for at danne eller vedligeholde nervernes myelinskede eller den aksonale struktur.
Der er på nuværende tidspunkt identificeret mere end 80 gener, som spiller en rolle i udviklingen af CMT, og der findes stadig flere. Generne er hver især forbundne med en særlig undertype.
CMT nedarves på forskellige måde, både autosomalt dominant, autosomalt recessiv og x-bundet.
-
Symptomdebut kan være fra barnealderen og op i voksenalderen. Symptomerne er mangeartede, og selv inden for den samme familie kan der være stor forskel på, hvordan og hvor hurtigt sygdommene udvikler sig.
De første symptomer er nedsat muskelkraft i fødder og underben. Det viser sig ved høj svang og vrist og krummede tæer (hulfod). Fodballen bliver specielt fremstående og kan blive øm, når man går.
Svag muskelkraft i fodleddet kan ses som dropfod. Desuden ses kluntet, trippende gang og usikkerhed ved at gå på ujævnt underlag. Nogle personer har problemer med at stå stille, da den nedsatte muskelkraft giver ustabile ankelled.
Der kan være nedsat muskelkraft i hænderne. Symptomerne ses ved svind af musklerne oven på hænderne, som kan føre til fejlstillinger i hænderne.
Kraftnedsættelsen kan progrediere til musklerne i underbenet, og der kan opstå udtalt svind af muskler i underbenene, som beskrives som “omvendt champagneflaske-atrofi”, også kaldet “storkeben”. Det vil sige meget tynde underben og kraftige lår. Man kan desuden få problemer med balancen på grund af den nedsatte muskelkraft i ankelleddet.
Følesansen (den taktile sans) kan være påvirket. Det kan blandt andet betyde, at man har svært ved at mærke varme og kulde. Der er øget risiko for sårdannelse på fødderne pga. uerkendt tryk fra fodtøj. Hos nogle personer med CMT kan følesansen være forøget, så det kan være smertefuldt at blive berørt bestemte steder.
Stillingssansen (den proprioceptive sans) kan også blive påvirket af sygdommen. Det kan være svært at vide, hvordan fødderne er placeret, uden at man samtidig ser på dem. Det kan give problemer med at holde balancen og færdes i mørke.
Ved nogle typer af sygdommen forekommer også hørenedsættelse.
Sygdommene er ofte langsomt progredierende.
Nogle vil i voksenlivet få behov for en elektrisk kørestol, når de færdes udendørs, men kun få personer vil få brug for kørestol permanent.
Almindelige komplikationer
Som følge af sygdomsudviklingen kan der opstå en række komplikationer:
- Forstuvninger af anklerne på grund af de ustabile ankelled
- Knogle- og ledforandringer i fødder, knæ, hofter og hænder
- Træthed både fysisk og psykisk – som blandt andet kan forklares ved, at man bruger mange kræfter på at udføre et arbejde, som for andre ikke kræver så meget
- Smerter – som kan opstå uden forudgående belastning og beskrives som jagende, krampeagtige smerter
- Kuldefornemmelser – en “dyb” kulde, specielt i fødderne
- Tremor – det vil sige, at hænder og fødder ryster i hvile og ved bevægelse.
- I sjældne tilfælde kan der opstå rygskævhed (skoliose), eller der kan komme nedsat vejrtrækningsevne på grund af nedsat kraft i vejrtrækningsmusklerne.
Personer med CMT forventes at have en levealder svarende til normalbefolkningen.
Undertyper
Type 1
Denne type er den hyppigste form og udgør ca. 2/3 af tilfældene. CMT1 er karakteriseret ved, at der sker en demyelinisering (nervens fedtskede svinder). CMT1 har en autosomal dominant arvegang.
Der ses distal muskelsvaghed og atrofi i både under og overekstremiteter. Der kan ses hyppige ankelforstuvninger, samt påvirkning af de sensoriske nerver, som kan medføre paræstesi og usikker, kluntet gang. Symptomerne starter ofte allerede i barndommen.
-
CMT1A Skyldes en duplikation i PMP22 genet på kromosom 22. PMP22 er vigtigt i forbindelse med dannelse og vedligeholdelse af myelinskederne i det perifere nervesystem.
CMT1A er langt den hyppigste form for CMT og udgør ca. 70 % af CMT1. Symptomdebut er typisk i teenageårene eller i 20’erne, men man kan se symptomer tidligere end dette.
Den kliniske præsentation er klassisk den, der er beskrevet ovenfor i sygdomsforløb. Der ses kraftnedsættelse, atrofi og sensorisk påvirkning ofte startende i den distale muskulatur på ben/fødder og siden på hænder/arme. Typisk har personer med CMT1A været langsomme løbere i barndommen, de udvikler høj svang -hulfod og hammertæer. Der er varierende grad af påvirkning af hænderne, men også her kan udvikles betydelige fejlstillinger.
Der er oftest problemer med balancen og en del patienter rapporterer også om smerte og træthed.
De fleste forbliver gående og den forventede livslængde er normal
CMT1B er den næsthyppigste form for CMT1. CMT1B skyldes en mutation i MPZ-genet på kromosom 1.
Den kliniske præsentation af CMT1B minder meget om CMT1A, men der er en større variation med debut helt fra den tidlige barnealder til i 20`erne.
CMT1C er en sjælden form for CMT. Den skyldes en mutation I LITAF-genet der sidder på kromosom 16. Den kliniske præsentation er meget lig den for CMT 1A
CMT1D er en sjælden form for CMT, der udgør mindre end 1% af alle CMT undertyperne. CMT 1D skyldes en mutation I EGR2 på kromosom 10.
Klinisk viser sygdommen sig oftest allerede i de tidlige barneår med forsinket motorisk udvikling.
Ved nogle få tilfælde er der også beskrevet respiratoriske problemer.
CMT1E er en sjælden form, der skyldes punkt mutation i PMP22 genet på kromosom 17. Samme gen som også er involveret i andre neuropatier (CMT1A og HNPP). CMT1E har tidligere debut og alvorligere forløb end CMT1A. Der ses ofte symptomdebut inden for de første 2 år med forsinket motorisk udvikling.
Flere med CMT1E vil få brug for kørestol.
CMT1F Meget sjælden form. Skyldes mutation i NEFL-genet på kromosom 8.
Type 2
Kaldes også aksonal type, da det er aksonet, der afficeres. CMT 2 nedarves, for langt hovedparten af undertyperne, autosomalt dominant.
Den kliniske præsentation minder meget om type 1 med distal muskelsvaghed, muskelatrofi og sensorisk påvirkning.
Ved CMT2 ses en større spredning i debutalder og en større variation af funktionsnedsættelse.
Den hyppigste form for CMT2 er CMT2A2
-
CMT2A1 Skyldes en dominant arvelig mutation i KIF1B-genet på kromosom 1. Der ses debut i barnealderen med muskelsvaghed i de perifere muskler på benene.
CMT2A2 Historisk blev CMT2A associeret med mutation i KIF1B (se ovenstående beskrivelse for CMT2A1). Siden er det opstået tvivl om dette. Derfor tales der nu om CMT2A1 og CMT2A2. CMT2A2 skyldes en mutation i MFN2-genet på kromosom 1. Der findes både en dominant og en recessiv form, hvorfor man kan støde på betegnelsen CMT2A2A (dominant) og CMT2A2B (recessiv).
CMT2B skyldes en dominant arvelig mutation i RAB7-genet på kromosom 3. Klinisk adskiller CMT2B sig ved at have en større grad af sensorisk påvirkning og mindre motorisk påvirkning. Der beskrives desuden en øget tendens til distal sårdannelse/risiko for kroniske sår.
CMT2C Meget sjælden form for CMT, der skyldes en dominant arvelig mutation i TRPV4-genet på kromosom 12. Klinisk ses ved denne type af CMT også påvirkning af diagphragma og stemmebåndene.
CMT2D Skyldes dominant arvelig mutation i GARS genet på kromosom 7.
Det kliniske billede adskiller sig ved at nogle med CMT2D næsten udelukkende har motorisk påvirkning, mens andre har både sensorisk og motorisk påvirkning.
CMT2E Skyldes dominant arvelig mutation i NEFL-genet på kromosom 8.
Klinisk ses meget stor variation i debut alder og graden af påvirkning.
CMT2F Skyldes en dominant arvelig mutation i HSPB1-genet på kromosom 7. Mutationer i samme gen er også involveret i andre arvelig neuropatier. Der ses oftest debut i 20-40 årsalderen, og der vil ses mere udtalt kraftnedsættelse i den distale muskulatur i både arme og ben.
CMT2G er blevet omklassificeret til CMT2P
CMT2H er kun beskrevet i en slægt/familie. Er en recessiv arvelig CMT med aksonal påvirkning og også pyramidal påvirkning med bl.a. livlige reflekser i over- og underekstremiteter.
CMT2I Sjælden skyldes dominant arvelig mutation i MPZ-genet på kromosom 1. Klinisk ses senere debut (over 50 år), men kan efter debut være hurtigt progredierende.
CMT2J skyldes dominant arvelig mutation i MPZ-genet på kromosom 1. Ved CMT2J kan forekomme døvhed og påvirkning af pupillen.
CMT2K skyldes en mutation i GDAP1-genet. Den kan nedarves både dominant og recessivt. Der er stor variation i symptomdebut til før 10 årsalderen til efter 50 år.
CMT2L Sjælden form, der skyldes en dominant arvelig mutation i HSPB8-genet på kromosom 12.
CMT2M omdøbt til intermediær form for CMTB dominant arvelig. – DI CMTB. Skyldes en mutation på DNM2-genet på kromosom 19.
CMT2N skyldes en dominant mutation i AARS1-genet på kromosom 16. Sjælden form.
CMT2O skyldes en mutation i DYNC1H1-genet på kromosom 14. Andre mutationer i genet er forbundet med andre neuromuskulære sygdomme. Ved denne form for CMT ses næsten kun motorisk påvirkning og kun begrænset eller ingen sensorisk affektion.
Debut i barneårene med forsinket motorisk udvikling, påvirket gang og svært ved at løbe.
CMT2P skyldes en mutation i LRSAM1-genet på kromosom 9. Mutation kan nedarves både dominant og recessivt.
Klinisk beskrives meget forskellige forløb, men generelt langsomt progredierende og typisk debut i voksenårene. Der er beskrevet fascikulationer i ben, erektil dysfunktion samt forhøjelse i serum creatinin kinase (CK).
CMT2Q skyldes mutation i DHTKD1-genet på kromosom 10. Der er på nuværende tidspunkt kun beskrevet én familie i Kina med mutationen.
CMT2R Skyldes mutation i TRIM2 genet på kromosom 4. Sjælden og ikke beskrevet i Danmark, men der er beskrevet familier med mutationen i Finland og Tyrkiet.
CMT2S Skyldes mutation på IGHMP2 genet på kromosom 11. Nedarves recessivt. Mutationen i genet kan også give mere alvorlige neuromuskulære sygdomme (SMA RD1).
CMT2T Skyldes mutation på MME-genet på kromosom 3. Ikke beskrevet i Danmark. Klinisk beskrives langsomt progredierende CMT med debut i voksenalderen.
CMT2U Skyldes mutation i MARS 1 genet på kromosom 12. Ikke beskrevet i Danmark.
Klinisk beskrives sen voksen debut (40 -60 årsalderen) og kun langsom progression
CMT2V Skyldes dominant arvelig mutation i NAGLU-genet op kromosom 17. Meget sjælden, mutationen er kun beskrevet i en fransk- canadisk familie.
CMT2Z Skyldes en mutation i MORC2- genet på kromosom 22, mutationen nedarves dominant.
Klinisk er der beskrevet debut fra tidlig barndom til i voksenalderen. Som andre CMT typer starter sygdommen typisk mest distalt, men over årene progredier CMT2Z til også at afficere muskler over knæ og albuer – inkl. hofte, skuldre og nakkemuskulaturen.
Personer med CMT2Z får ofte brug for mobilitetshjælpemidler og de fleste anvender kørestol når man er i 50´erne.
Intermediær form
Er en sjælden form for CMT, og der er kun beskrevet få tilfælde i Danmark. Den beskrives som en blandingsform mellem CMT1 og CMT2. Ledningshastigheden er reduceret, men ikke i så høj grad som ved type 1.
Den Intermediære form er oftest dominant arvelig, men der findes også typer med recessiv arvegang.
Når undertyperne af den Intermediære form skal betegnes, bruges D-for dominant og R- for recessiv og I for intermediær. Herefter indsættes -CMT og så et bogstav for undertype f.eks. DI-CMTA
Type 4
Er en sjælden recessiv nedarvet form for CMT. Der er tale om en demyeliniserende type. Ved nogle typer af CMT4 er der også symptomer fra andre dele af kroppen som f.eks. gråstær og hørenedsættelse.
-
CMT 4A skyldes en mutation i GDAP1 genet på kromosom 8. Mutation i samme gen kan også give CMT2K.
Klinisk ses debut i barne- og ungdomsårene – de fleste med debut før 10 år. Der ses affektion af både over og underekstremitet med muskelsvaghed, atrofi og kontrakturer. Hos børn ses forsinket motorisk udvikling.
De fleste med CMT4A har behov for støtte ved gang og ved 30 års-alderen har omkring 75 % behov for kørestol.
Senere i sygdomsforløbet kan der udvikles hæshed/påvirket stemme.
Der er normal forventet levetid.
CMT 4B Der beskrives både en CMT4B-1 og CMT4B-2, der skyldes mutationer i henholdsvis MTMR2-genet og SBF2-genet begge på kromosom 11. Begge gener har betydning for, hvorledes myelinskeden foldes.
CMT type 4 B har debut i barnealderen og er mere progressiv. De fleste bliver kørestolbrugere, men tidspunktet kan variere fra teenage-årerne til senere i voksenlivet.
Der kan ses påvirkning af ansigtsmuskulaturen og diaphragma (mellemgulvet). Der er også beskrevet bulbære symptomer, og hørepåvirkning kan også forekomme.
CMT 4C skyldes en mutation i SH3TC2- genet på kromosom 5. Der ses stor variation i graden af påvirkning. CMT4C kan debutere i barndommen med sensorisk og motorisk påvirkning i især ben. Ved CMT 4C er der risiko for udvikling af scoliosis.
Ved denne type af CMT kan der også være høretab, påvirket stemme og påvirket ansigtsmuskulatur.
CMT 4D Skyldes mutation i NDRG1 genet på kromosom 8.
Kliniks ses distal kraftnedsættelse, muskelatrofi og sensorisk påvirkning, samt fejlstillinger af fødder og hænder.
Personer med CMT4D udvikler oftest høretab/døvhed i 30’erne.
CMT 4E skyldes en mutation i ERG2 på kromosom 11. Der er tale om en medfødt form for CMT og den omtales oftest som Congenital hypomyelinating neuropathy (CHN). https://omim.org/entry/605253
CMT 4F en sjælden form for recessiv CMT, der skyldes en mutation i PRX genet på kromosom 19.
CMT 4F en sjælden form for recessiv CMT, der skyldes en mutation i PRX genet på kromosom 19. Debut ses fra tidlig barndom til voksen med langsom progression af typisk CMT-fænotype med sensorisk og motorisk påvirkning især i ben. Forsinkede milepæle ved tidlig debut.
CMT 4G skyldes en mutation i HK1-genet på kromosom 10. Typen kaldes også the Russe type of hereditary motor and sensory neuropathy (HMSNR). Debut typisk inden for de første 10 leveår. Det kliniske billede svarer til det, der er beskrevet under sygdommens forløb ovenfor.
CMT 4H Skyldes mutation i FGD4-genet på kromosom 12. Det er en sjælden form, der kun er beskrevet i 4 familier. Den kliniske præsentation varierer, men som beskrevet oven for under ”sygdommens forløb”.
CMT 4J Skyldes mutation i FIG4- genet på kromosom 6. Sjælden form for CMT, der først blev beskrevet I 2007. Da der kun er få beskrivelser, kan der ikke sige noget entydigt om den kliniske præsentation, men de fleste har klassiske symptomer som beskrevet oven for under ”sygdommens forløb).
Type X
CMTX er en kønsbunden arvelig form. Ved denne type svinder nervetråden (aksonal form), men der ses også samtidig forandringer i fedtskeden. Der er beskrevet 6 undertyper. CMTX1 er den hyppigste form af de X-bundne former af CMT og udgør mellem 10-15 % af alle CMT.
-
CMT X1 eller CMT1X Den næsthyppigste form for CMT, som udgør mellem 10-15 %. Skyldes mutation i GJB1 genet på X-kromosomet. Da kvinder har 2 X-kromosomer og dermed også 2 GJB1 gener er der en tendens til, at kvinder rammes mildere i forhold til mænd i samme familie. Men kvinderne med sygdomsgenet har også symptomer.
CMT X2 Meget sjælden form, der nedarves X-bundet recessivt, dvs. at kvinder kan være bærere af sygdommen uden selv at have symptomer.
CMT X3 Nedarves X-bundet recessivt, dvs. at kvinder kan være bærere af sygdomsgenet uden selv at have symptomer.
CMT X4 med eller uden ataksi, kaldes også Cowchock syndrom (COWCK). Typen skyldes mutation i AIFM1 genet på X-kromosomet. Sygdomsgenet nedarves recessivt. Mutationer i samme gen kan også give X-bundet døvhed.
CMT X4 kan være med eller uden cerebellar ataksi. Klinisk kan der være debut fra tidligste barndom til ung-voksen alder. Det kliniske udtryk kan variere meget, men der kan være forsinket motorisk udvikling, høretab/døvhed og påvirket gang.
Der er også beskrevet kognitiv påvirkning, cerebellar atrofi ved hjerneskanning, cerbellare symptomer som dysartri, tremor , dysmetri (kan fx ikke ramme et mål med finger) og spasticitet. Der ses store variationer inden for samme familie.
CMT X5 Skyldes mutation i PRPS1-genet, og samme mutation kan også være skyld i flere syndrom sygdomme (Arts syndrom og X-bundet døvhed). Der ses et stort fænotypisk (det kliniske udtryk) overlap mellem syndromerne og CMTX5, hvorfor det diskuteres om CMTX5 er en selvstændig sygdom eller mere en del af et syndrom. Man kan betragte de 3 sygdomme som repræsenterende et fænotypisk spektrum med samme genetiske baggrund. For alle 3 sygdomme gælder, at der er en tendens til at mænd rammes hårdere end kvinder.
CMT X6 Skyldes mutation i PDK3 genet, der nedarves dominant. Mutationen er på nuværende tidspunkt kun beskrevet i én familie. Da der kun er få beskrivelser, kan der ikke sige noget entydigt om den kliniske præsentation, men de fleste har klassiske symptomer som beskrevet oven for under ”sygdommens forløb”.
Behandling
Medicin: På nuværende tidspunkt findes der ikke nogen medicin, der kan helbrede sygdommen. Der forskes meget i behandling, men typisk er der tale om behandling målrettet en enkelt undertype, og ikke hele gruppen af CMT.
Foddeformiteter: Kan ofte afhjælpes med specialfodtøj individuelt tilrettet ved ortopædist. Ved mere udtalte deformiteter bør patienten henvises til ortopædkirurg med speciel viden om CMT (højt specialiseret niveau). Behandlingen kan indebære operation med flytning af muskelsener, artrodese, osteotomi, etc.
Smerter: Kan behandles med lettere analgetika, hvis smerterne vurderes udløst fra bevægeapparatet. Ved neurogene smerter behandles efter vanlige principper herfor.
Fysioterapi: Patienter med CMT bør tilrådes fysisk træning med henblik på bedring muskelstyrke og balance. Patienter kan henvises til vederlagsfri fysioterapi.
RCFM har oversat en guide til fysioterapi, som primært henvender sig til personer med CMT, men som også indeholder viden for fagpersoner. I guiden kan du læse, hvad en person med CMT kan forvente af sin fysio- og ergoterapeut, hvordan terapien kan afhjælpe nogle af de problemer, der opstår med sygdommen, og hvilke former for hjælpemidler og træning, der anbefales.
Hjælpemidler: Har til hensigt at understøtte eller kompensere for påvirkede funktioner.
Særlige opmærkomhedspunkter
Medicin: Da visse typer medicin kan påvirke de perifere nerver, er det vigtigt altid at overveje dette inden der ordineres medicin. Der anbefales særligt stor forsigtighed med præparater, der har kendt neurotoksitet. Overvej, om der findes et alternativ. Hvis ordinerende læge er i tvivl, anbefales det at konferere med kontrollerende neurolog.
Den amerikanske CMT-association har en medicinliste med præparater, man skal være særlig opmærksom på.
RCFM har i forbindelse med udbruddet af COVID-19 udarbejdet et akutkort, hvor der også er beskrevet særlige hensyn
Genetisk rådgivning: Patienter og relevante familiemedlemmer bør tilbydes genetisk rådgivning, som kan foregå enten på neurologisk afdeling eller på klinisk genetisk afdeling afhængigt af problematikken.
Graviditet: Patienterne kan, hvis muligt, tilbydes prænatal diagnostik ved graviditet efter forudgående rådgivning og genetisk undersøgelse. Patienter med diagnosen, der påtænker graviditet skal være opmærksom på, at der kan være visse komplikationer og risici forbundet med graviditet.
Forsikring: Diagnosen CMT vil i mange tilfælde kunne udløse forsikringssum ved kritisk sygdom, derfor anbefales det at personer, der får stillet diagnosen, undersøger dette med sit forsikringsselskab.


