
Progressiv muskelatrofi (PMA)
Progressiv muskelatrofi (PMA) er en sjælden sygdom og en af de 4 undertyper af ALS-sygdommen.
Læs mere om MND nederst på siden.
Sygdommen rammer 2.neuron, dvs. nervecellerne i hjernestammens kerner og forhornscellerne i rygmarven, som leder nerveimpulset ud til skeletmusklerne i henholdsvis ansigt, tunge, svælg, muskelgrupper i arme, ben, og vejrtrækningsmusklerne.
Påvirkning af 2. neuron giver symptomer som pareser (muskelslaphed), svage senereflekser, muskel atrofi (tab af muskelmasse), reduceret muskelkraft og fascikulationer (muskelsitren).

Der ses ikke påvirkning af de sensoriske nervebaner og følesansen er bevaret. Synssansen og høresansen forbliver ligeledes intakt gennem sygdomsforløbet.
-
Forekomst
Som andre former for MND (link til MND sygdomsbeskrivelse) rammes typisk personer over 50 år. PMA rammer lidt flere mænd end kvinder.
Der er ca. 20 personer med PMA tilknyttet RCFM
Årsag
Der forskes i de underliggende sygdomsmekanismer, men indtil videre kendes de ikke. Der er formentlig tale om multifaktorielle årsager, hvor både genetik og udefrakommende påvirkninger spiller ind.
-
PMA-diagnosen kan i starten være meget vanskelig at stille. Det skyldes, at der ikke er et enkelt sygdomstegn eller en enkelt undersøgelse, der giver en entydig diagnosticering af sygdommen. Derfor har nogle personer med PMA været igennem et langt udredningsforløb, inden de får stillet diagnosen.
PMA-diagnosen stilles, ved at neurologen kan påvise 2.neurons symptomer, som der ikke kan forklares ved anden sygdom. Samtidig må der ikke være påvirkning af følesansen eller andre sanser.
PMA er en klinisk diagnose med forværring over tid. For at udelukke andre sygdomme i nervesystemet med lignende symptomatologi, kan der foretages en række undersøgelser, som kan understøtte PMA-diagnosen eller afkræfte denne.
- Blodprøver: foretages rutinemæssigt.
- Genetisk undersøgelse: kan være relevant, hvis der har været flere tilfælde i familien.
- Rygmarvsvæskeundersøgelse: kan udelukke andre årsager og være med til at påvise 1. neurons påvirkning.
- MR- eller CT-scanning af hjerne og rygmarv: Kan udelukke andre diagnoser, hvor tryk på nervebaner kan forekomme.
- Neurofysiologisk undersøgelse i form af EMG (elektromyografi), ENG (elektroneurografi) og MEP (motor evoked potential) og SSEP (Somato-Sensorisk-Evokerede-Potentialer). Ved EMG måles den elektriske aktivitet i musklerne, der ved MND viser et bestemt mønster med denervering og evt. Ved ENG måles nerveledningshastigheder i de motoriske nerver (2.neuron), og ved MEP måles, hvordan 1. neuron fungerer. Ved SSEP er formålet at undersøge de sensoriske nervebaner. Den neurofysiologiske undersøgelse kan være med til at understøtte den objektive neurologiske undersøgelse.
-
Sygdommen starter ofte i arme eller ben medførerende nedsat muskelkraft, atrofi, krampetendens og synlige fascikulationer. Der ses ofte vægttab. Sygdommen udvikler sig meget forskelligt fra person til person, men der vil efter en periode (op til år) ses spredning til andre dele af kroppen. Symptomerne er jævnt progredierende både i sværhedsgrad og i antallet af involverede områder.
-
PMA udvikler sig forskelligt fra person til person, og en del med patienter med PMA lever i mere end 5 år.
Sygdommen progredierer over tid, og der vil være et stigende behov for hjælp til alle funktioner. Respirationen vil efterhånden blive tiltagende svækket, og der vil være stigende risiko for lungeinfektioner. I takt med at respirationsmusklerne svækkes, vil der ske en gradvis CO2-ophobning, der i sidste ende medfører at patienten bliver tiltagende komatøs og til sidst sover ind.
Læs mere om den sidste tid her
-
Medicin: Der findes ingen helbredende medicinsk behandling. Den behandlingen, der gives, har til sigte at mindske følgevirkningerne fra sygdommen.
Der kan fx gives medicinsk behandling med muskelafslappende midler i forbindelse med krampetendens.
Fysioterapi: Det er vigtigt at foretage udspænding for at bevare mobiliteten og undgå fejlstilling af inaktive led og muskler. Fysisk aktivitet kan dæmpe eventuelle muskelkramper. Træning af de ikke-afficerede muskler er ligeledes vigtigt, for at undgå overbelastningsskader samt med henblik på at bevare et så højt funktionsniveau som muligt.
Læs de kliniske retningslinjer for fysioterapi til patienter med ALS.
Ergoterapi: til hjælp ved dysfagi og i forhold til hjælpemidler.
Logopædisk behandling: ved eventuelle dysartri-gener
Palliative teams: Når sygdommen er fremskreden, og der kommer et tiltagende behov for lindrende behandling, anbefales det at inddrage de palliative specialister.
Læs mere om den sidste tid her
Relevante samarbejdspartnere
ALS team på neurologiske afdelinger
Respirationscentre
Palliative specialister
Kommunale støtteforanstaltninger
Kommunikationscentre
Behandlende fysioterapeut
Referencer:
https://www.mndassociation.org/
Beskrivelse af motorneuronsygdom (MND)
ALS (Amyotrofisk lateral sklerose) er en sygdom, der er lokaliseret i de motoriske nerveceller, og de benævnes derfor overordnet motorisk nervesygdomme.
Den engelske betegnelse MND (Motor Neuron Disease) benyttes mere og mere, også i Danmark og af de danske neurologer. Betegnelsen MND dækker over en række karakteristiske, progredierende symptomer og kan inddeles i 4 hovedtyper. Sammensætningen af symptomer samt rækkefølgen de opstår i, kan være afgørende for, hvilken type MND der diagnosticeres.
De 4 hovedtyper er:
- Klassisk ALS
- Progressiv bulbær parese (Bulbær ALS)
- Progressiv muskelatrofi (PMA)
- ALS med Fronto Temporal Demens eller frontotemporal dysfunction (ALS/FTD eller ALS/FTDdys)
Fælles for de 4 typer er påvirkningen af de motoriske neuroner (nerveceller), der ved viljens hjælp styrer musklernes bevægelse, og som i takt med sygdommens progression gradvist går til grunde.
Hvilken af de 4 typer, der er tale om, afhænger af, hvilke motoriske neuroner der er påvirket. Man inddeler de motoriske neuroner i 1.neuron (øvre), og 2. neuron (nedre).
Første neuron betegner de motoriske neuroner, der ligger i den motoriske cortex (hjernebarken), hvor axonet (nervetråden) løber ned til et nyt motorisk neuron (2. neuron) beliggende enten i medulla oblongata (forlængede marv), eller i medulla spinalis (rygmarvens) forreste del, kaldet forhornet (2.neuronet i forhornet benævnes også forhornscelle).
Andet neuron sender et langt axon ud til skeletmuskulaturen i hele kroppen, herunder også respirationsmuskler (vejrtrækningsmuskler).
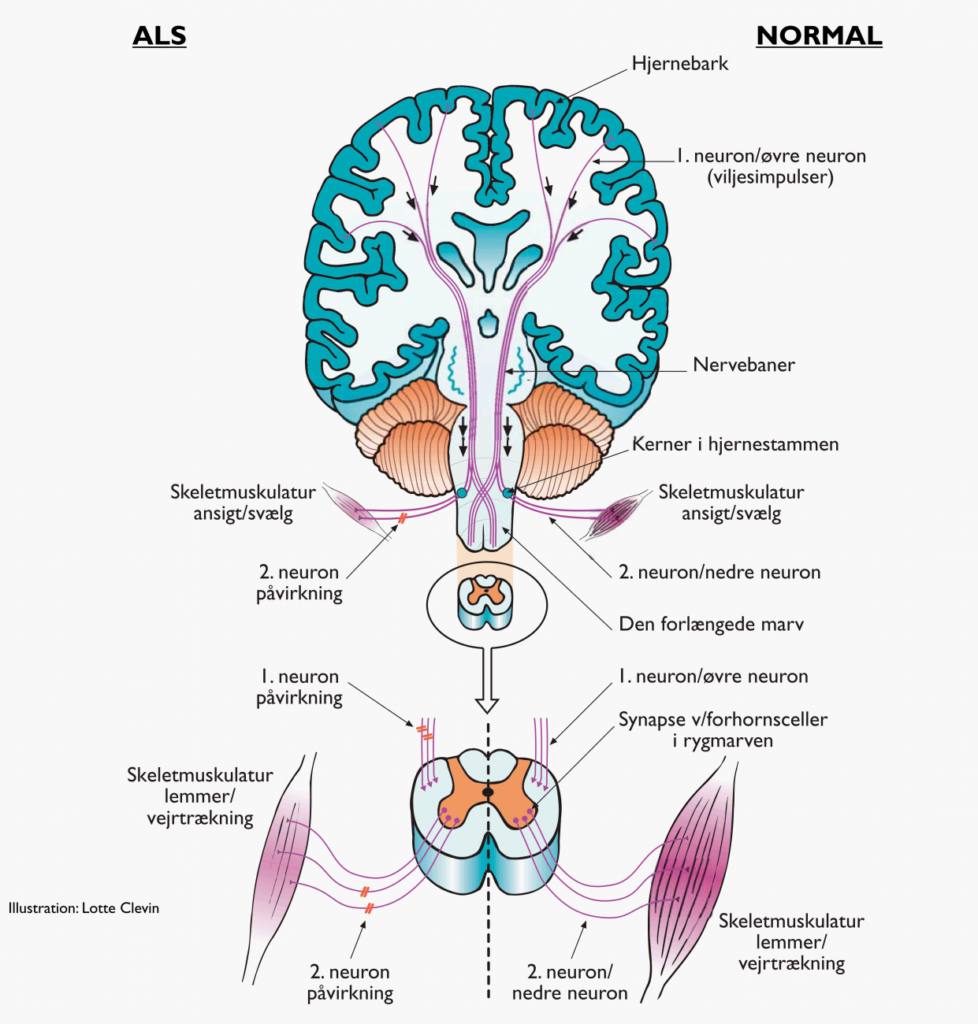
Ved klassisk ALS, Bulbær ALS og ALS/FTD er både 1. og 2. motorneuron påvirket i mindst en region af kroppen. Ved PMA er der alene påvirkning af 2. motoriske neuron i mindst to regioner af kroppen. For nærmere beskrivelse af symptomer ved de enkelte hovedtyper, se venligst sygdomsbeskrivelse for disse
Ødelæggelsen af neuronerne og axonerne kan således opstå og udvikle sig i hjernen og/eller i rygmarven. Afhængig af hvor påvirkningen starter, kan symptomerne vise sig i fx en arm eller svælget, eller mere udbredt i fx begge arme, eller arme og ben. Efterfølgende kan symptomerne sprede sig til resten af kroppens skeletmuskulatur. Sygdomsforløbet for den enkelte kan derfor være meget forskelligt, afhængigt af hvordan og hvor neurodegenerationen (nerveødelæggelse) og dermed symptomerne starter.
Tidligt i sygdomsforløbet kan det være vanskeligt at skelne mellem de forskellige typer, da der er overlappende symptomer.
Inddelingen af ALS-sygdommen i de 4 undertyper er et forsøg på at beskrive sygdommens forventede udvikling og prognose på diagnosetidspunktet. Det er vigtigt at bemærke, at nogle som først diagnosticeres som PMA senere i forløbet kan få ændret diagnosen til ALS i takt med at deres symptomer udvikler sig.


