
Oversigt over forskning i DMD
Formålet med oversigten er at informere patienter med Duchenne og deres familier om de forskellige behandlingsmuligheder, der forskes i lige nu. Artiklen beskriver fordele og ulemper ved de forskellige behandlingsmetoder samt hvilke forhindringer, der skal overvindes, inden behandlingerne kan bruges af patienterne.
Det videnskabelige felt inden for behandling af Duchenne er kæmpestort og i hastig udvikling, og dette gør det umuligt at beskrive alle forskningsprojekter i en kort oversigt. Det følgende er derfor en beskrivelse af hovedtrækkene i forskningen.
Problemet bag Duchenne, som skal løses
Duchenne muskeldystrofi skyldes mutationer (genetiske fejl), der ødelægger den genetiske kode for dystrofingenet.
Genet producerer det dystrofinprotein, der forbinder skelettet af muskelfibrene til det beskyttende lag på ydersiden af fibrene. Denne forbindelse stabiliserer muskelfibrene, når musklerne trækker sig sammen (træning). Dystrofinets funktion kan sammenlignes med et anker (musklens skelet) og en båd (musklens beskyttende lag), hvor dystrofinet er det reb, der forbinder anker og båd.
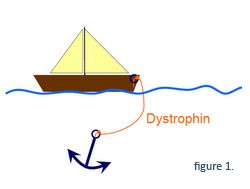
På grund af mutationen på den genetiske kode, kan cellen kun producere begyndelsen af proteinet, og forbindelsesfunktionen er væk (båden er ikke længere forbundet til ankeret, Figur 2). Resultatet er, at muskelfibrene ved Duchenne nemt beskadiges, selv ved almindelig træning.

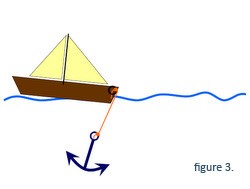
Mutationer i dystrofingenet, der ikke helt ødelægger den genetiske kode, tillader dannelsen af et dystrofin, der er delvist funktionelt (dvs. det kan forbinde ankeret til båden, men kæden er lidt kortere, Figur 3). Disse mutationer forbindes med den knapt så alvorlige Beckers muskeldystrofi.
Lige nu udvikles der på mange medicinske behandlinger, og størstedelen af dem følger en skabelon, hvor der først laves prækliniske og derefter kliniske forsøg. I prækliniske forsøg testes behandlingen i dyrkede patientceller, derefter på dyremodeller med sygdommen (normalt på mus). Først når resultaterne i celler og dyremodeller er tilstrækkeligt overbevisende, testes behandlingen på patienter. Dette er de kliniske forsøg, og denne del af udviklingen kan tage meget lang tid, da der let opstår uforudsete problemer, der skal løses. Kliniske forsøg er opdelt i flere faser: I fase 1, udføres forsøget for at påvise, om behandlingen er sikker. I fase 2 og 3 er målet at finde ud af, om behandlingen er både sikker og effektiv. Det er en god huskeregel, at selv om hver fase er kritisk, er den næste fase altid mere udfordrende. Så selv om et produkt ser lovende ud i et præklinisk forsøg (fx i et cellestudie), er der ingen garanti for, at det også virker i videre forsøg på dyr og mennesker.
Mulige løsninger
Overordnet findes der fire metoder til at forbedre kroppens muskler, når man har Duchenne.
- Genterapi
- Stamcelleterapi
- Behandling med medicin
- Metoder målrettet den enkelte mutation
Nedenfor gennemgås, hvordan man udvikler behandlinger på baggrund af metoderne og hvilke forsøg, der er i gang.
Genterapi
Kliniske forsøg
-
Et amerikansk/canadisk fase 3-forsøg med genterapien RGX-202, har nået sit primære mål. Resultaterne vækker forsigtig optimisme, men skal stadig skal bekræftes i flere forsøg. Resultaterne viste at:
- 93 % af de behandlede drenge opnåede mindst 10 % af det normale dystrofinniveau – målet for studiet.
- 80 % nåede over 40 %, og gennemsnittet lå på 71 %.
- Det dannede mikrodystrofin fandtes i muskelcellernes membran, hvor det normalt skal virke.
- Foreløbige data tyder på forbedret motorisk funktion sammenlignet med ubehandlede patienter – især hos dem med høj proteinproduktion.
- Behandlingen blev generelt tålt godt. Bivirkninger var hovedsageligt milde (fx kvalme og træthed). To alvorlige tilfælde blev behandlet uden varige mén.
En vigtig pointe er, at studiet fandt en klar sammenhæng mellem mængden af mikrodystrofin og forbedringer i funktion
Regenxbio, som har udviklet medicinen vil søge om såkaldt accelerated approval hos FDA. Det kan give tidligere adgang til behandling.
En bekræftende undersøgelse er allerede i gang og skal dokumentere de langsigtede effekter. Hvis alt går som planlagt, kan behandlingen blive tilgængelig i USA i 2027.
-
Medicinalvirksomheden Genethon har udviklet en genterapien GNT0004.
Virksomheden oplyser, at tre drenge med Duchenne muskeldystrofi, som har fået den genterapeutiske behandling GNT0004 i et klinisk forsøg, stadig har bedre motorisk funktion to år efter de fik behandlingen
Behandlingen er udviklet til at levere et gen til muskelcellerne, som kan få dem til at producere mikrodystrofin – en forkortet, men funktionel version af dystrofinproteinet – med det formål at bevare musklerne og bremse sygdommens progression. Genet føres ind i cellerne ved hjælp af et virus, der er omprogrammeret til at levere et terapeutisk gen i stedet for at give infektioner.
Fase 3-forsøg påbegyndt i efteråret 2025
Et forsøg med samme dosis GNT0004 testes nu på 64 drenge i alderen 6 til 10 år, som stadig kan gå. Forsøgene finder sted i Frankrig og England.
Resultater fra de tre GNT0004-behandlede drenge
Resultaterne fra de tre patienter, der fik GNT0004, blev sammenlignet med data fra drenge, som ikke havde fået behandlingen.
Drengene i forsøget blev testet med North Star Ambulatory Assessment, som bedømmer motorisk funktion på en skala op til 34. En forbedring på mere end 2,5 point anses for klinisk relevant. Efter 18 måneder var den gennemsnitlige score for de behandlede drenge 5,8 point højere end i kontrolgruppen. Drengene viste også forbedringer på tiden det tager at rejse sig fra gulvet eller gå en kort distance, og disse forbedringer er opretholdt i op til to år.
Endelig var niveauerne for creatinphosphokinase (CPK), en biomarkør for muskelskade, reduceret med 75 % efter 18 måneder og 61 % efter to år. Målinger af musklens fedtfraktion, som sig noget om, hvor meget musklen er nedbrudt, viste også forbedringer, og der blev ikke rapporteret om alvorlige bivirkninger, oplyser Genethon.
-
Om Elvidys
Elevidys er den første godkendte genterapi mod Duchenne muskeldystrofi i USA, men er endnu ikke godkendt i Europa
Medicinen har siden juni 2023 været godkendt til mindre børn af det amerikanske FDA, og i 2024 blev godkendelsen udvidet til at omfatte alle med Duchenne fra 4 år og op. Børn som har deletioner i exon 8 og/eller exon 9 kan af sikkerhedsmæssige grunde ikke få Elevidys. Elevidys er en engangsbehandling, der gives intravenøst.
EMA har siden juni 2024 været i gang med at behandle en ansøgning om godkendelse af Elevidys, men er endnu ikke nået til en afgørelse.
Usikkerhed om Elevidys efter to dødsfald
Elevidys kan have bivirkninger i form af leverskade, og i henholdsvis marts og juni 2025 døde to personer, som var behandlet med Elevidys af leversvigt. Begge var kørestolsbrugere.
Siden marts 2025 er alle forsøg med Elevidys i Europa sat på pause, som følge af et påbud fra det europæiske medicinagentur EMA.
I juli 2025 besluttede EMA ikke at godkende Elevidys til markedsføring.
Efter det andet dødsfald er medicinen til kørestolsbrugere trukket tilbage i de lande, hvor den er godkendt, mens gående stadig kan få behandling.
Sarepta, som har udviklet Elevidys, arbejder nu på at lave en ny sikkerhedsprotokol, der kan gøre det mere sikkert at få medicinen.
Kliniske forsøg
Resultaterne fra de første forsøg med Elevidys (EMBARK) nåede ikke målsætningen om statistisk signifikant forbedring af funktionsniveau målt med North Star- skalaen. Til gengæld havde medicinen signifikant effekt på nogle sekundære funktionelle slutmål, bl.a. tid til at rejse sig fra gulvet og 10-meters gangtest.
I USA følger man de behandlede patienter fra EMBARK-forsøget. En analyse fra januar 2026 viser, at de efter tre år stadig rejser sig ca. 6 sekunder hurtigere fra gulvet og går 10 meter 2,7 sekunder hurtigere end ikke-behandlede patienter.
-
Pfizer har i juni 2024 afbrudt et fase3-studie med minidystrofin-genterapi på drenge mellem 4 og 7 år. Årsagen er, ifølge Pfizer, at målet om en forbedring i motorisk funktion ikke blev indfriet. Et år efter påbegyndt behandling kunne der ikke måles forberinger med North Star Ambulatory Assessment-skalaen, ej heller kun man måle forskel på gruppen, der fik medicin, og placebo-gruppen ved en 10 meter gangtest eller tid til at rejse sig fra gulvet.
Forskning i genterapi
Ved at indsætte et sundt gen i Duchenne-musklerne forsøger man at gøre det muligt at producere mere dystrofin.
Gener består af DNA og findes på kromosonerne, som befinder sig i cellernes kerne. Dystrofingenet indeholder den genetiske kode, som kan sammenlignes med opskriften på dystrofin. Koden læses af cellen og oversættes til produktion af dystrofinprotein.
Udfordringer og løsninger
At få det raske gen ind i alle muskelcellerne
Musklerne udgør en stor del af kroppen. Ca. 30-40 % af kroppens vægt er muskler. Kroppen består af mere end 750 forskellige muskler, der hver består af flere milliarder celler, og det raske gen skal derfor indsættes i en meget stor del af cellekernerne i alle musklerne.
Heldigvis findes der en organisme, der er rigtig god til at føre gener ind i cellerne, nemlig virus. Inden for genterapien har man derfor udviklet virusser, hvor det sygdomsfremkaldende virusgen er fjernet for at give plads til det raske gen. Ved hjælp af en sådan virus (kaldet en viral vektor) forsøger man, at få det raske gen ind i muskelcellerne.
At finde en effektiv virus
De fleste virusser kan godt lide at inficere celler, der deler sig. Muskelvæv deler sig næsten ikke, og er derfor ikke et typisk mål for virus. Derudover er muskelfibrene svøbt ind i flere lag bindevæv, der opfanger viruspartikler, og virus kan derfor ikke nå ind til muskelfibrene med dystrofingenet.
Der findes en virus, som er relativt god til at inficere muskelceller, den såkaldte AAV-virus. Denne virus kan inficere menneskeceller, men er ikke sygdomsfremkaldende.
At skabe en virus, der er lille nok
Desværre er AAV så lille, at dens genetiske kode for dystrofin ikke passer (hele genet er ca. 500 gange for stort, den genetiske kode ca. 4 gange for stor).
Forskerne har forsøgt at skabe det mindst mulige dystrofin, hvis genetiske kode er så lille, at det kan være i en AAV-virus.
I forsøg med at behandle mus med mikrodystrofin med AAV-virusser fik musene bedre muskelkvalitet og muskelfunktion.
At undertrykke kroppens eget immunforsvar
I en hundemodel (golden retriever med muskeldystrofi), der blev behandlet med AAV-mikrodystrofin, reagerede immunforsvaret ved at ødelægge de celler, der var inficeret med AAV-mikrodystrofin. Fra kliniske forsøg på mennesker med andre sygdomme (fx blødersygdom) ved vi også, at AAV aktiverer immunforsvaret. Immunforsvaret angriber alle ubudne gæster (virusser, bakterier, parasitter), og den ved ikke, om den angriber en sygdomsfremkaldende virus eller en virus, der bærer på et gavnligt gen.
Dette kan løses ved at give høje doser binyrebarkhormon (kortekosteroider).
Behandling med medicin
Standardbehandling med kortikosterioder (binyrebarkhormon)
Kortikosteroider (også kaldet binyrebarkhormon) er en gruppe medicinske præparater, der kan undertrykke immunforsvaret. Når muskelcellerne går i stykker, opfatter kroppen det som om, der er en inflammation.
Kroppens reaktion er at skrue op for immunforsvaret. Det bekæmper inflammationen, men det har også den uheldige effekt, at der frigives stoffer, der ødelægger cellerne. Ved at undertrykke immunforsvaret med kortikosteroider bliver beskadigelsen af musklerne ikke så slem, og der dannes mindre bindevæv.
Der er almindelig enighed om, at kortikosteroider forhaler sygdomsudviklingen. Derfor er steroidbehandling standardbehandling for Duchenne.
Behandlingen
- udsætter afhængighed af kørestol med ca. 1-3 år,
- er med til midlertidigt at forbedre muskelstyrke og -funktion,
- forsinker tab af vejrtrækningsfunktion.
Steroidbehandling har ikke været anvendt længe nok til, at man ved, om den er livsforlængende. Forskning viser, at steroidbehandling sandsynligvis også har andre fordele end at undertrykke immunforsvaret (man mener, at de kan øge udskillelsen af utrofin og/eller stabilisere muskelfibrene, så de er mindre sårbare over for beskadigelse). Der forskes stadig i dette.
Bivirkninger
Kortikosteroider skal tages regelmæssigt og kontinuerligt. Det giver bivirkninger hos de fleste patienter. De mest almindelige er vægtøgning, depression, adfærdsproblemer, forsinket vækst, forsinket pubertet og tab af knoglemasse, men mange flere er beskrevet.
Hos nogle patienter kan bivirkningerne nedbringes ved kun at tage medicinen i perioder, fx kun hver anden uge eller i ugedagene og ikke i weekender, eller kun høje doser i weekenderne. Nogle patienter har færre bivirkninger med præparatet Deflazacort.
Hvis bivirkningerne er større end fordelene (fx vægtøgning i en sådan grad, at det svækker musklernes funktion i stedet for at øge den), kan det være bedst at stoppe behandlingen. Dette bør selvfølgelig kun gøres efter konsultation med den behandlende læge, da brat afbrydelse af steroidbehandling kan give alvorlige bivirkninger.
Tidspunkt for opstart af behandling
Man ved ikke, hvornår det er bedst at starte behandling med kortikosteroider. På grund af bivirkninger (væksthæmning, fedme og forøget knogleskørhed) starter de fleste læger først behandlingen op i 3-4 års-alderen.
For at teste, om det er gavnligt at starte inden 3-årsalderen, er der lige nu et forsøg i gang på drenge mellem 1 og 30 gamle. De får en høj dosis prednison, men kun to gange om ugen for at mindske risikoen for bivirkninger.
Prednison og Deflazacort
Disse to lægemidler har længe været givet som standardbehandling i Danmark. De menes at have samme virkning og være omtrent lige gode. Forskellige læger og hospitaler kan foreskrive forskellige doser og intervaller for behandlingen. Det kan være nødvendigt at justere dosis, skifte præparat og iværksættes andre tiltag og medicin for at mindske uønskede bivirkninger.
Kliniske forsøg med forskellige typer medicin
Normalt kan medicin tages gennem munden og virke på alle kroppens muskler (der er ikke problemer med få den ind i muskelcellerne som ved gen- eller celleterapi). Nogle gange kan medicin, som anvendes til behandling af andre sygdomme, bruges til at behandle Duchenne-patienter. Dette gør det hurtigere at afprøve medicinen på mennesker, da sikkerhed og dosis allerede er testet.
-
Vamorolone – en steroidlignende behandling med færre bivirkninger
Vamorolone er et ikke-steroid-præparat udviklet af Reveragen Biopharma. Stoffet har samme gavnlige virkning som steroidbehandling, men uden nogle af de alvorlige bivirkninger.
Resultaterne af et forsøg over 48 uger, kaldet VISION-DMD, viste at vamorolone gav en forbedring af den fysiske funktion, men uden at påvirke væksten. I et open-label forsøg med vamorolone, som har fundet sted over en periode på 2,5 år, blev der ikke rapporteret om hæmmet vækst, samtidig med at der blev rapporteret om færre bivirkninger. Også i VISION-DMD-forsøget sås det, at de drenge, der fik 6 mg/kg/dag fortsatte med at vokse, hvilket ikke var tilfældet med dem, der fik prednison.
Vomorolone blev godkendt til behandling i EU i december 2023. Behandlingen sælges under navnet AGAMREE, men er endnu ikke godkendt til Danmark. En ansøgning behandles lige nu af Medicinrådet, som forventes at nå en beslutning i december 2025.
Forsøg med Satralizumab
Satralizumab er en antiinflammatorisk behandling, der virker ved at blokere et inflammatorisk signalmolekyle kaldet interleukin-6 (IL-6). Satralizumab er godkendt under navnet Enspryng til behandling af den autoimmune sygdom neuromyelitis optica.
Et nyt forsøg skal teste, om Satralizumab også kan være med til at kan forebygge knogleskørhed og øge eller bevare muskelfunktionen.
Rekrutterer deltagere til fase 2-forsøg
De kommende forsøg, som er åben for rekruttering, foregår USA, Italien, Polen og Spanien
Forsøget er et fase 2-forsøg kaldet SHIELD DMD (NCT06450639), hvor behandlingen testes på gående og ikke-gående drenge med DMD i alderen 8 til 15 år.
I forsøgene bliver deltagerne inddelt i to grupper: gående deltagere, som tidligere har haft brud på knoglerne og ikke-gående deltagere med eller uden tidligere brud (Gruppe 1) samt gående deltagere, der ikke tidligere har haft brud (Gruppe 2) ved studiestart.
Deltagerne får en dosis, som er beregnet ud fra deres vægt hver 4. uge. Behandlingen gives gennem indsprøjtning under huden (subkutan injektion)
Patienter, der får exon-skipping- eller genbehandlinger, kan ikke deltage.
Sikkerhed og bivirkninger
Tidligere forsøg har vist, at behandlingen er sikker, og almindelige bivirkninger er hovedpine, ledsmerter, lavt antal hvide blodlegemer og reaktioner på injektionsstedet.
-
Når man har DMD er stofskiftet i muskelcellerne nedsat. Det skyldes bl.a., at mitokondrierne, som er cellernes energi-centraler, ikke kan danne så meget energi. Ved at give medicin, der kan styrke mitokondrierne, håber man at kunne forbedre muskelfunktionen.
Klinisk forsøg
Epicatechin er et hormonllignende molekyle, som dannes af mitokondrierne efter fysisk udfoldelse. Mitokondrier er cellernes kraftværk, der leverer energi til cellerne. Epicatechin har vist sig at øge produktionen af mitokondrier og forbedre gendannelse af musklerne samt reducere bindevævsdannelse. Et mindre pilotstudie på patienter med Beckers muskeldystrofi har vist gode resultater og at epicatechin var veltolereret. Et forsøg med ikke-gående patienter med Duchenne er netop afsluttet hos producenten Cardero Therapeutics. Resultaterne er endnu ikke opgjort.
Ifetroban har vist sig at reducere inflammation og arvæv, især i hjertet, ved andre diagnoser. Cumberland Therapeutics undersøger derfor, om lægemidlet også kan anvendes til Duchenne-patienter.
-
Når en muskel trækker sig sammen, skubbes blod ud af blodkarrene, selv om den sammentrukne muskel faktisk har brug for mere blod (ilt og næringsstoffer). Uden dystrofin kan blodkarrene i hjerte og muskler ikke udvides tilstrækkeligt. Dette kan medføre, at der ikke kommer ilt nok til muskler og hjerte, hvilket igen kan beskadige muskel- og hjertecellerne. Der findes mange godkendte præparater, der kan forbedre udvidelsen af blodkarrene. De præparater, der er opført nedenfor, testes nu eller er testet på Duchenne-patienter i kliniske forsøg.
Lisinopril
Lisinoprils virkning på hjertefunktionen testes nu på patienter med Duchenne i USA, Japan og Canada. Disse forsøg undersøger også, om samtidig brug af Coenzym Q10 (en antioxidant) og lisinopril yderligere har gavnlig indvirkning på hjertemusklens funktion. Der er endnu ikke kommet resultater af forsøgene.
-
HDAC-inhibitorer
I takt med sygdommens fremadskriden dannes der mere og mere bindevæv i musklerne hos en person med DMD. Ved at give medicin, der hæmmer dannelsen af bindevæv, håber man at kunne bedre muskelfunktionen og sinke progressionen i sygdommen. Denne type medicin kaldes HDAC-inhibitorer.
Givinostat godkendt af FDA
Givinostat er en HDAC-inhibitor, som er sikker til brug for børn. Den er nu også testet på personer med Duchenne i en italiensk undersøgelse. Resultaterne fra det første lille forsøg viste, at behandling i et år blev tålt i rimelig grad. Analyse af muskelbiopsier tydede på, at bindevæv og fedt var reduceret sammenlignet med biopsier taget før og efter forsøgsstart. Et opfølgende forsøg, hvor alle får medicinen, er nu i gang på 5. år. Analyser tyder på, at personer, der får medicinen, bevarer gangfunktionen længere, end personer, der ikke får den.
Et internationalt fase 3-forsøg med gående personer med DMD, viste, at dem, der blev behandlet i 72 uger, klarede sig signifikant bedre i en trappetest end gruppen, der fik placebo. De klarede sig også bedre i 6 minutters gangtest, North Star-testen og i tidsfunktionstests. MR-analyse viste reduceret udvikling af fedt i musklen.
På baggrund af disse resultater har medicinalvirksomheden Italfarmaco indsendt en ansøgning om markedsføringstilladelse til Duchenne-patienter hos FDA og EMA.
FDA godkendte i marts 2024 givinostat, som i handlen kommer til at hedde Duvyzat, til Duchenne-patienter i alderen 6 år og ældre. EMA gav en betinget godkendelse af Duvyzat i juni 2025. At godkendelsen er betinget betyder, at patienter får adgang til medicinen, men at EMA vil se resultater fra flere kliniske forsøg, inden de giver en endelig godkendelse. Italfarmaco har også ansøgt Medicinrådet om godkendelse og der forventes en afgørelse sidst i oktober 2025.
Læs mere om, hvordan Duvyzat virker her
Og se en forklaring for børn her
Myostatin-hæmmere
Når man har Duchenne, går muskelcellerne langsomt til grunde, uden kroppen kan skabe nye til erstatning for de ødelagte. I vores krop findes et protein, myostatin, der har den virkning, at det forhindrer musklerne i at vokse for meget. Det er nyttigt, fordi det fx kan bremse kræftcellers vækst. Dette har man forsøgt at udnytte i behandlingen. Ved at give medicin der svækker myostatinens virkning, håber man, at kroppen bliver bedre til at producere nye muskelceller og dermed modarbejde det tab af muskelceller, som Duchenne medfører. Der er foretaget en række kliniske forsøg med myostatinhæmmere, men ingen af dem vist de ønskede resultater. Follistatin gen-levering
Follistatin er et protein, der hæmmer myostatin. Som beskrevet ovenfor er myostatin et protein, der hæmmer musklernes vækst. Så ved at forøge mængden af follistatin, hæmmes hæmmeren, hvilket gør det muligt for muskelmassen at vokse. Follistatin-genet er blevet leveret til aber og mus ved hjælp af en AAV viral vektor. Indsprøjtningerne resulterede i øget muskelmasse og muskelstyrke.
Et klinisk forsøg, hvor AAV-virus med follistatin-genet indsprøjtes i lårmusklen på Becker-patienter er i gang i USA. Formålet er at undersøge, om stoffet er sikkert, og om det kan forbedre muskelmassen og -styrken i lårmusklen. I et opfølgende forsøg er stoffet nu også testet på Duchenne-patienter, og foreløbige resultater viser, at det er veltolereret.
Normalisering af kroppens kalciumbalance
På grund af manglende dystrofin bliver kalciumkanalen i muskelfibrene utæt, og det medfører unormalt store mængder kalcium i musklerne. Dette resulterer i muskelskader, oxidativ stress og bindevæv. Præparater, der kaldes ‘Rycalls’ kan normalisere kalciumbalancen, fordi de kan korrigere lækagen. Behandlingen har vist sig gavnlig på mus. Der udføres i øjeblikket et klinisk forsøg på Duchenne-patienter.
Et andet potentielt præparat, der kan normalisere kalciumindholdet i muskelfibrene, hedder rimeporide. Præparatet er testet sikkert og der planlægges nu et fase-2-forsøg.
Beskyttelse af muskelfibrene
Manglende dystrophin gør muskelfibre følsomme over for skader under sammentrækning. Dette gælder især for fibre med en stor diameter. Edgewise udvikler en oral forbindelse (EDG5506), der kan beskytte de mest skadesfølsomme muskelfibre. Denne forbindelse testes i øjeblikket på raske frivillige og patienter med Becker muskeldystrofi samt på Duchenne-patienter.
-
Lige nu screenes tusindvis af medicinske præparater for om man kan finde et, der kan øge mængden af utrofin.
Utrofin er et protein, der ligner dystrofin meget og danner den samme forbindelse mellem celleskelettet og bindevævet som dystrofin, men hovedsageligt i andre vævstyper end muskelvæv. Under opbygning af musklerne befinder utrofin sig i muskelfibrenes membran. Når produktionen af dystrofin begynder, vil dystrofinet imidlertid erstatte utrofinet.
I voksne muskler findes utrofin kun i meget lave mængder og hovedsageligt ved forbindelsen mellem nerve og muskel (den neuromuskulære forbindelse). I Duchenne-patienter og dyremodeller findes utrofin dog også i muskelfibermembranen, men hos patienterne er disse mængder for små til at gøre nogen gavn. Studier på mus har vist, at højere mængder utrofin kan kompensere for den manglende dystrofin på funktionsniveau og også forsinke sygdomsudviklingen.
Kliniske forsøg
Der er foretaget kliniske forsøg med to præparater, men desværre gav ingen af den de ønskede resultater.
To andre præparater med at øge utrofin-niveauet testes nu i den prekliniske fase.
Behandling med stamceller
Kliniske forsøg med stamceller
-
Hjertestamceller har måske potentiale til at forsinke udviklingen af hjerteproblemer hos personer med DMD.
Biotekvirksomheden Capricor Therapeutics har udviklet en stamcellebehandling, Deramiocel, tidligere CAP-1002.
Et fase 1-forsøg med drenge og unge med Duchenne fra 10 år og op, som havde hjerteproblemer, viste at behandlingen var veltolereret, og der sås en lille reduktion af fibrose (arvæv) i hjertet. Forsøget viste også tegn på øget styrke i skulder- og armmusklerne, hvilket kunne tyde på, at stamcellebehandlingen også kunne have en positiv effekt på skeletmuskulaturen.
Et fase-2-forsøg bekræftede behandlingens positive virkning på både hjerte og muskler, og dette er nu yderligere bekræftet i et fase tre-forsøg.
Forsøget inkluderede 106 drenge og mænd med DMD over 10 år, som tilfældigt blev tildelt enten deramiocel eller placebo hver tredje måned. De fleste deltagere kunne ikke gå, og mere end tre fjerdedele havde kardiomyopati.
Deltagerne blev fulgt i et år og blev testet for arm- og håndfunktion samt hjertefunktion. Resultaterne viste en næsten 54 procents opbremsning af sygdomsprogression i skeletmuskulaturen og en 91 procents opbremsning af sygdomsprogression i hjertemuskulaturen.
Capricor Therapeutics håber, at resultaterne rækker til at få medicinen godkendt af de amerikanske sundhedsmyndigheder.
Forskning i stamceller
Ved at overføre muskelceller med et rask gen fra raske donorer til Duchenne-muskler forsøger forskerne at kompensere for manglende muskelvæv og at give donorcellerne mulighed for at starte en normal produktion af dystrofin.
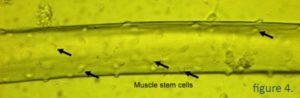
Muskelfibre og muskelstamceller
Muskler består af muskelfibre, der ikke deler sig, og muskelstamceller, der ligger oven på fibrene (Figur 4). Når muskelfibrene er beskadiget, begynder muskelstamcellerne (også kaldet satellitceller eller myoblaster) at dele sig og bevæge sig hen til det beskadigede sted, hvor de smelter sammen med den beskadigede muskel for at reparere den. Muskelstamceller kan isoleres fra en muskelbiopsi og dyrkes i et laboratorium derefter transplanteres til en Duchenne-muskel.
Udfordringer og løsninger
Muskelstamceller kan ikke bevæge sig fra blodbanen og ind i musklen. Løsningen på dette er at give en lokal injektion i den syge muskel.
Selv hvis stamcellen indsprøjtes direkte ind i musklen, bevæger den sig dog ikke mere end 1-2 mm fra det sted nålen stikkes ind. Derfor vil man skulle foretage flere indsprøjtninger (fx 100 pr. cm2). Dette er afprøvet på Duchenne-patienter, hvorefter celler med dystrofin har kunnet ses på injektionsstedet.
Klinisk forsøg med indsprøjtning af et højt antal stamceller
I Canada indsprøjtede man muskelstamceller med 100 injektioner på et lille område (0.25-1 cm2). Behandlingen var uden alvorlige bivirkninger, og man fandt dystrofinpositive fibre i en biopsi taget fra det behandlede område. I Canada afprøver man lige nu lokal transplantation af muskelstamceller i en underarmsmuskel på patienter på 16 år og derover.
Desværre er det er ikke muligt at anvende denne metode til at levere muskelceller til alle kroppens muskler. Heldigvis findes der andre stamceller i blodet, i blodkarrenes vægge og i fedtvævet, som også kan medvirke til muskeldannelse. Disse celler kan isoleres og dyrkes i et laboratorium. Fordelen ved dette er, at cellerne sandsynligvis kan bevæge sig fra blodbanen ind i musklerne og derved muliggøre behandling af hele kroppen.
Kun lille effekt
Selv om sådanne celler kan medvirke til muskeldannelse, er effekten på nuværende tidspunkt meget lille (mindre end 1 % af den transplanterede celle ender i musklerne).
I Italien har man netop gennemført et forsøg, hvor celler blev taget fra Duchenne-patienter (isoleret fra blodet), dyrket i laboratoriet og derefter transplanteret tilbage i patienternes muskler.
Der forskes lige nu i, hvordan man kan øge effekten af denne metode. Der er opnået lovende resultater i muse- og hundemodeller med forskellige typer stamceller.
Celler fra raske søskende
I Italien har man gennemført et klinisk forsøg for at teste sikkerheden ved at transplantere celler fra raske brødre til Duchenne-patienter. Da det kun var en sikkerhedstest, fandt man ikke forbedring af muskelfunktionen, men arbejder videre med at forbedre metoden.
Omprogrammering af celler
Det er svært at skaffe tilstrækkelige mængder stamceller til transplantation, fordi stamcellerne ikke er gode til at formere sig.
Derfor har man fundet ud af at omprogramere voksne celler, så de får nogle af de samme egenskaber som stamceller. Disse celler er rigtig gode til at formere sig og kan omdanne sig til alle mulige celletyper. Men udfordringen er at få dem til kun at omdanne sig til muskelceller. Der arbejdes lige nu forbedre metoden.
Muskeltransplantation
Transplantation af donormuskler vil aktivere immunforsvaret (som ved transplantation af enhver type væv til en anden person).
Når man modtager donervæv er det nødvendigt at give medicin, der undertrykker immunforsvaret. Desværre er kronisk behandling med denne type medicin ikke uden bivirkninger (fx at man er mere modtagelig for infektioner).
I stedet forsøger man at isolere muskelceller fra patienterne, dyrke dem i laboratoriet og behandle dem (fx med genterapi). Derefter transplanteres patienternes egne celler tilbage (kaldet autolog transplantation). Genterapi er meget mere effektivt på celler (i laboratoriet) end i væv (i en person). Derudover kan man måske undgå brug af medicin, der undertrykker immunforsvaret, fordi det er patientens egne celler, der transplanteres.
Kroppen kan reagere på manipulerede celler
For at dette skal virke, er man nødt til at optimere de måder, hvorpå muskelceller eller andre stamceller kan leveres effektivt. Immunforsvaret kan også stadig reagere på de transplanterede celler, selv om de er fra patienten selv, fordi de er blevet manipuleret i laboratoriet og derved sandsynligvis har ændret sig.
I laboratoriet er det nu muligt at foretage mindre ændringer i cellens DNA uden at skulle tilføje et gen. Dette sker ved hjælp af såkaldte DNA-”sakse”. Arbejdet med disse DNA-sakse er vanskeligt. I dyrkede celler, skal den celle, hvor saksen har haft succes, først findes (normalt kun ca. 1 ud af 1000), derefter dyrkes for til sidst at blive transplanteret. Tre forsøg på mus har vist gode tegn, men udfordringen ved genterapi og celleterapi er at oversætte metoden fra musestadiet til større dyr og mennesker.
Behandling målrettet den enkelte mutation
Exon skipping og gennemlæsning af “stop codons”
Exon skipping og gennemlæsning af ”stop codons” er en type behandling, der virker for bestemte genmutationer. Dette betyder, at den kun virker på den del af patienterne, som har særlige mutationer (se de følgende sider for mere information). For at finde ud af, om en Duchenne-patient er egnet til behandling med exon skipping eller gennemlæsning af ”stop codon”, er det vigtigt at kende den fulde genetiske diagnose (dvs., at den mutation, der forårsager sygdommen i dystrofingenet, skal være identificeret).
-
Godkendte lægemidler, der bruger exon skipping (ikke godkendt i EU)
I øjeblikket er der fire lægemidler, der er godkendt af FDA, nemlig eteplirsen (exon 51), golodirsen (exon 53), casimersen (exon 45) and viltolarsen (exon 53). I Japan er lægemidlet viltolarsen godkendt. Alle produkterne har opnået en såkaldt accelereret godkendelse på baggrund af deres evne til at producere små mængder dystrofinprotein. Hvorvidtså små mængder dystrofinprotein har nogen indvirkning på sygdomsprogressionen forskes der i lige nu. Ingen af lægemidlerne er godkendt i EU.
Forskning i Exon skipping
Med Exon Skipping er det muligt at korrigere den genetiske kode og tillade produktionen af delvist funktionelt dystrofin.
Den genetiske kode er splittet ud på såkaldte exons. Når der skal dannes et protein, laver genet en midlertidig kopi (kaldet RNA). Inden dette RNA kan oversættes til protein, skal exons først samles, og de indskudte dele, der ikke indeholder genetisk kode (introns), skal slettes. Denne proces kaldes “splejsning”.
Hos Duchenne-patienter, er dystrofinets genetiske kode afbrudt, hvilket betyder, at koden bliver ulæselig, og derved afbrydes oversættelsen af genet til protein.Hos patienter med Beckers muskeldystrofi, som er en mildere version af Duchenne, har mutationerne beholdt den genetisk kode, hvilket gør det muligt at danne et funktionsdueligt protein.

Exon skippings formål er at genoprette den genetiske kode hos Duchenne-patienter, så der kan dannes et delvist funktionelt, Becker-lignende dystrofinprotein i stedet for et ikke-funktionelt Duchenne-protein.
Dette kan ske ved hjælp af såkaldte AON’er (antisense oligonucleotider), som er små stykker modificeret RNA, der genkender et mål-exon, binder sig til det og gemmer det fra splejsningsmaskineriet. Dette resulterer i en genoprettelse af den genetiske kode. I dyrkede celler fra patienter og i mdx-musemodeller er det lykkedes at lave exon skipping med AON-behandling, hvilket har skabt en produktion af Becker-lignende dystrofin. Exon 51 skipping
Eftersom skipping af exon nr. 51 er det, der kan anvendes på den største gruppe patienter (ca. 13 %), er man længst fremme i forskningen med at udvikle AON’er, der er målrettet exon 51. Et AON-præparat kaldet eteplirsen (markedsført som Exondys 51) er godkendt til behandling i USA men ikke i Europa. Eteplirsen gives ved indsprøjtning. Kliniske forsøg har vist, at eteplirsen giver en lille forøgelse af indholdet af dystrofin. Derudover viste et studie fra 2023 af såkaldte real-world data fra 579 patienter i behandling med Exondys 51, at patienter, der fik Exondys 51 i levede længere sammenlignet med naturhistorien.
Virksomheden PepGen udvikler på en lignende behandling kaldet PGN-EDO51. Et fase 2-forsøg er netop startet op. PepGen anvender en teknik, som de kalder Enhanced Delivery Oligonucleotide, til at få PGN-EDO51 ind i musklerne. Virksomheden mener selv, at denne teknik giver en mere effektiv exon-skipping end andre tilgange og derved i sidste ende resulterer i en højere produktion af dystrofin.
Exon 53 skipping
Golodirsen er ligesom eteplirsen et AON-præparat, der kan skippe exon 53. Golodirsen er godkendt til markedsføring i USA under navnet Vyondys 53. I et forsøg gennemført på 25 patienter i Europa målte man en “statistisk signifikant stigning” i produktionen af dystrofin i de skeletale muskler. FDA vurderede, at denne stigning, “med rimelig sandsynlighed kan give en klinisk fordel” for patienter med denne mutation.
Data fra patienter, der er behandlet med Golodirsen i seks år, viser, at behandlingen kan udsætte tab af gangfunktion i gennemsnitligt 2,4 år, og der er også tegn på, at medicinen kan bremse tab af lungefunktion.
Kliniske forsøg med AON’er målrettet andre exoner
Sarepta har derfor indledt et placebo-kontrolleret fase 3-studie over 96 uger, som har til formål at evaluere AON’er for exon 45 og 53 (casimersen). Foreløbige resultater fra muskelbiopsier viser øgede mængder dystrofin fra 0,9 % til 1.7 %. På baggrund af disse resultater godkendte FDA præparatet i februar 2021, men med besked om, at der stadig skal arbejdes på at dokumentere medicinens virkning på musklernes funktion.
Nippon Shinyaku (Japan) og NS-Pharma er i gang med et klinisk forsøg med exon 53 skipping (viltolarsen) i Japan og på gående patienter i USA. Efter 24 ugers behandling med høje doser (40 og 80 mg/kg) sås op til 5 % dystrofin i en muskelbiopsi. FDA godkendte præparatet i august 2020.
Andre præparater til skipping af exon 44, 52, 54 og 55 er i gang, og der er mange medicinalvirksomheder, der satser på exon skipping.
Kliniske forsøg med stoffet drisapersen er stoppet.
-
Behandling med præparater til gennemlæsning af ”stop codons”
Disse typer medicin virker kun på patienter med en “stop codon”-mutation. Sådanne mutationer påvirker ikke den genetiske kode, men indfører et stopsignal i midten af genet samt i slutningen af genet, hvor det tilkendegives, at oversættelsen af proteinet er fuldendt. Dette gælder for ca. 10-15 % af alle Duchenne-patienter.
Hvad er stop codons
Alle gener har et startsignal og et stopsignal, så maskineriet, der oversætter generne til proteiner, ved, hvor det skal begynde og stoppe. Nogle gange kan en lille mutation indføre et stopsignal inde i genet (ud over det, der findes i slutningen). Normale stopsignaler er normalt lidt anderledes end de muterede stopsignaler ((en sammenligning kan være et stopsignal i et trafikeret vejkryds (normalt) og et stopsignal på en motorvej (muteret)). Uanset hvad, vil cellerne følge stopsignalet og stoppe oversættelsen af proteinet for tidligt.
Man forsøger derfor at udvikle et lægemiddel, der kan tvinge cellerne til at ignorere det muterede stop codon og producere et helt dystrofinprotein.
Kliniske forsøg
Ved at screene et stort antal medicinske præparater har man fundet et præparat, ataluren, der kan tvinge cellerne til at ignorere de muterede stop codons uden giftige bivirkninger. Præparat sælges under navnet Translarna™ og er udviklet af PTC Therapeutics (USA). Det er et pulver, som skal blandes op og drikkes.
Præparatet fik en betinget godkendelse af EMA, men opfølgende forsøg viste ikke tilstrækkelige resultater, og i januar 2024 meddelte EMA, at de ikke ville forlænge godkendelsen.
Læs også
-

Forskning i Danmark
Forskere på Aarhus Universitetshospital udvikler behandling med stamceller til Duchenne muskeldystrofi
Læs mere